|
Dear all,
I figured out what the problem was or rather that there was no
problem at all.
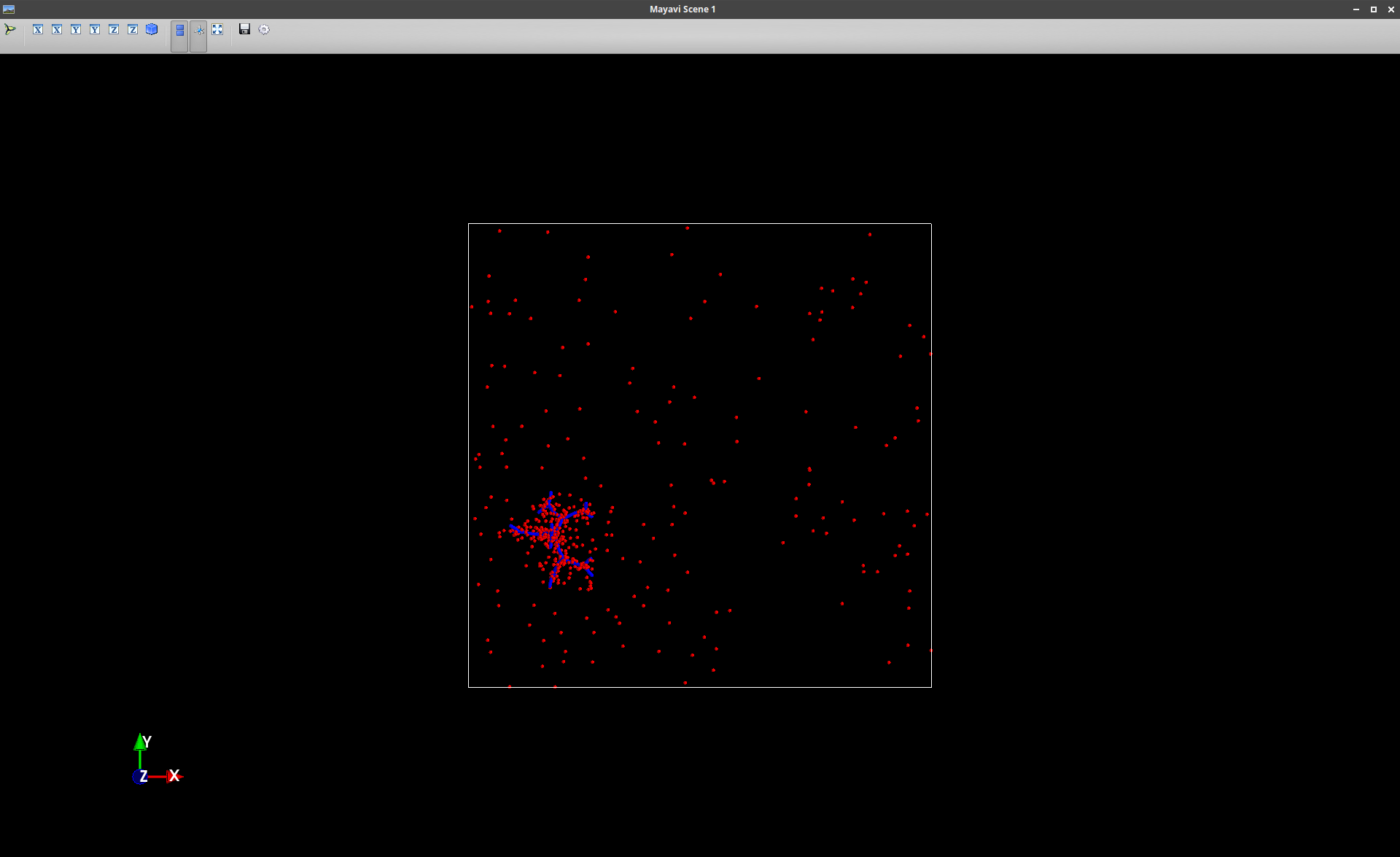
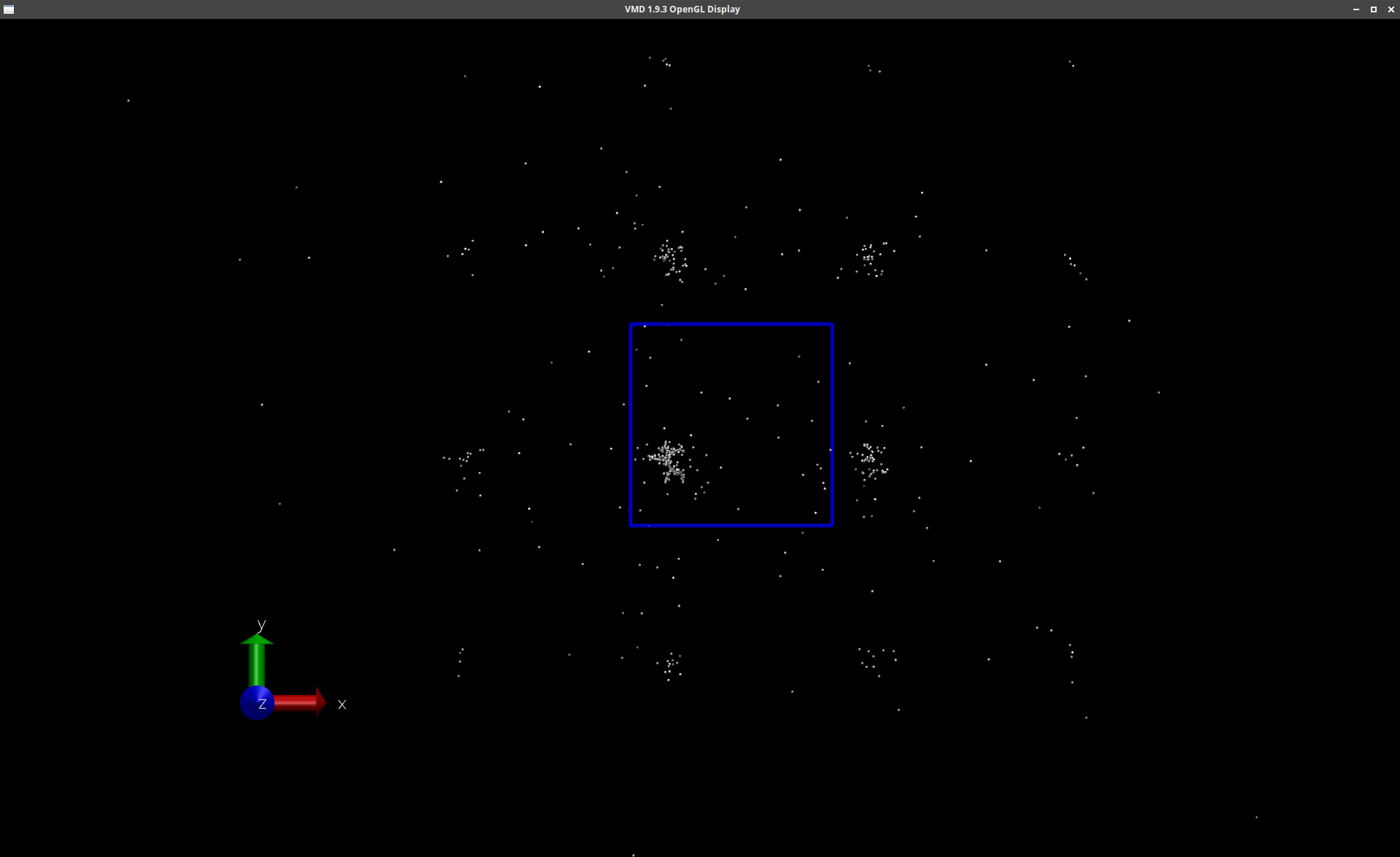
I decided to use a different tool for visualization and noticed
that VMD not only shows the original simulation box but also the
adjacent PBC boxes. For some reason only a part of the atoms is
visualized in these additional boxes. And strangely the
dendrimer is only shown in the original simulation box. So the
clustered counter-ions around the dendrimer from the original
box seemed to form clusters on their own in the additional PBC
boxes. I attached some screenshots.
Thanks for your all your help and advice!
Best,
Clemens
On 01.06.2017 15:10, Florian Weik
wrote:
Hello Clemens,
1) The error in P3M is not translation invariant (only on
average), so in principle it can cause such effects. If you
want to find out you can move the particle relative to the
mesh an see if anything changes. I would recommend you to use
a slightly larger mesh (maybe 32), and see if the effect goes
away. The tuning result looks odd to me, this could be a bug
in the tuning.go
As Rudolf was pointing out, you can fix the mesh and still
tune the other parameters, simply by providing a mesh (p3m
tune mesh 32 32 32 accuracy ...).
2) During the tuning process, the runtime of the force
calculation is measured. The warning means that the variance
of the samples was untypically big, this usually means that
there
that something else on the machine is interfering with your
espresso process. This can lead to a non-optimal tuning
results but has no influence otherwise. This is an effect of
the environment and not of the P3M parameters.
Otherwise, what Rudolf is saying.
Cheers,
Florian
Hi
Clemens,
On Wed, May 24, 2017 at 02:34:30PM +0200, Clemens Jochum
wrote:
> > I'm not sure I understnad your setup.
> > I suppose, you have two particle tyeps:
> > * particles making up the dendrimer (say type 0)
> > * salt ions (type 1)
> >
> > Now, there would be the following lj interactions:
> > dendrimer-dendrimer (0,0):
> > either sigma=bond length, epsilon=few kT,
cut_off=Something between 2^1/6 bond length and more,
depending on whether you want an attractive tail.
> > or cutoff=bond_length sigma=cutoff /(2^(1/6)),
in which case the bonds ould not be stretched by the lj
potential
>
> To clarify:
>
> The units in the dendrimer are polymer-like chains of
several monomers
> (see attached snapshot). The monomers in these chains
are bound by a
> harmonic bond with l_b = 3.4. I also have a harmonic
angle bond, which
> accounts for the stiffness of the chains. The
LJ-interaction is not
> needed for the interaction of neighbouring monomers,
but for the
> interaction between the chain-like arms of the
dendrimer.
>
> The parameters of the LJ interaction are:
>
> sigma = 4
> r_off = r_mon + r_mon - sigma = 14
> r_cut = 2^1/6 * sigma
>
> So it is a shifted WCA-potential that looks like:
>
> 4 * epsilon * (sigma / (r - r_off))^12 - (sigma / (r -
r_off))^6 if
> r_off < r < r_off + r_cut
>
> and it is 0 otherwise.
>
> Because the harmonic bond forces neighbouring monomers
to be around the
> point of divergence (r = r_off = 14 = 4.12 * l_b) of
the LJ-potential I
> encountered some problems. This is why I want to
exclude the 4 nearest
> neighbours from the LJ-interaction.
It is my impression that this is a rather non-standard
interaction setup. My suggestion would be to setup the
system with interactions as described in my previous mail,
i.e., on a monomer-monomer basis rather than on an arm-arm
basis. Once that system behaves as expected, you can re-add
more complexity. In this way, it should become clear, where
something goes wrong.
Regarding the correctness of the P3M method: Results for
several electrostatics methods agree
(testsuite/python/coulomb_cloud_wall.py). There is of
course, no guarantee that this hold for all situations, but
it would not be the first place, I'd look.
Regards, Rudolf
|
{kind=link}
{kind=link}